 /
/ 
Time Limit: 2 sec / Memory Limit: 1024 MiB
配点 : 500 点
問題文
sとtは文字
S_{sop} = \sum_{i=1}^{N} sim(s[i], t[i])
ただし,Nは
| x \ y | |||||
| 1 | -3 | -3 | -3 | -5 | |
| -3 | 1 | -3 | -3 | -5 | |
| -3 | -3 | 1 | -3 | -5 | |
| -3 | -3 | -3 | 1 | -5 | |
| -5 | -5 | -5 | -5 | -5 |
例えば,s=
S_{sop} = sim(
のように計算することができ,S_{sop}が最大となる.
この時,
C-TCGGT CATCC--
このように,文字列中の適切な場所に空白
二本の入力文字列についてS_{sop}を最大化するペアワイズアラインメントを出力せよ.
制約
- sとtは
A ,C ,G ,T からなる文字列である. - 10 \leq |s|, |t| \leq 400 (文字列xの長さを|x|と記述する.)
入力
入力は以下の形式で標準入力から与えられる.
s t
出力
入力文字列のS_{sop}が最大となるペアワイズアラインメントを出力せよ.例に倣い,sとtの適切な位置に
入力例 1
AGTTGAATTT GTCGGACTTT
出力例 1
AGT-TGAATTT -GTCGGACTTT
問題の背景
※ この項目は読まなくても問題を解くことができますが,出題の背景となりますため,ご興味を持ってくださった方はお読みいただけると幸いです.
ペアワイズアラインメントはゲノム配列解析の様々な場面において用いられます.ここでは,個人のゲノム配列解析について考えてみたいと思います.
DNAからゲノム配列を読み出す装置をシークエンサーと呼びます.ヒトのゲノム配列は全長30億塩基に及ぶ非常に長い配列ですが,シークエンサーはこの長い配列を端から端まで一気に読み出せるわけではありません.装置によって読み出しの長さは異なりますが,短いもので数百,長いもので数百万塩基程度の断片を読み出すのです.ですから,読み取った断片配列がゲノム上のどの部分に相当するのか突き止めなければなりません.そこで,既に決定されているヒトの標準的なゲノム配列(参照ゲノム配列と呼ぶ)と見比べて,読み取った断片配列の位置を特定するのです.ここで注意しなければならないのは,読み出した断片配列とそれに対応する参照ゲノムの部分配列には違いがあるという点です.差異が生じる理由は二つあります.一つは,ゲノム配列は個人(個体)ごとに少しずつ異なるためで,もう一つはシークエンサーがゲノム配列を読み出す際にエラーを発生させてしまうためです.これら二つに共通するのは,参照ゲノム配列に対して次のような差異が比較的多く生じるということです.
置換(ある文字が別の文字に置き換わっている.)
ATGC ← 参照ゲノム配列 ATCC ← 読み出した個人のゲノム配列
欠損(ある文字が欠損している.)
ATGC ← 参照ゲノム配列 AT-C ← 読み出した個人のゲノム配列
挿入(ある文字が追加されている.)
ATC-C ← 参照ゲノム配列 ATCAC ← 読み出した個人のゲノム配列
このような差異のある配列同士の類似部分をうまく並べるために,ペアワイズアラインメントが必要となるのです.
なお,ゲノム配列全長とシークエンサーで読み出した断片配列のように長さが大きく違う配列をアラインメントするには様々な工夫が必要となります.また,良いアラインメントを作るためには類似度の設計も重要です.例えば,挿入があまり入らないと予想されるケースでは
シークエンサーについて
さて,上の文章で書いた「個人のゲノム配列解析」が気軽にできるようになったのは,実はごく最近のことなのです.次のグラフをご覧ください.(NIHより引用)
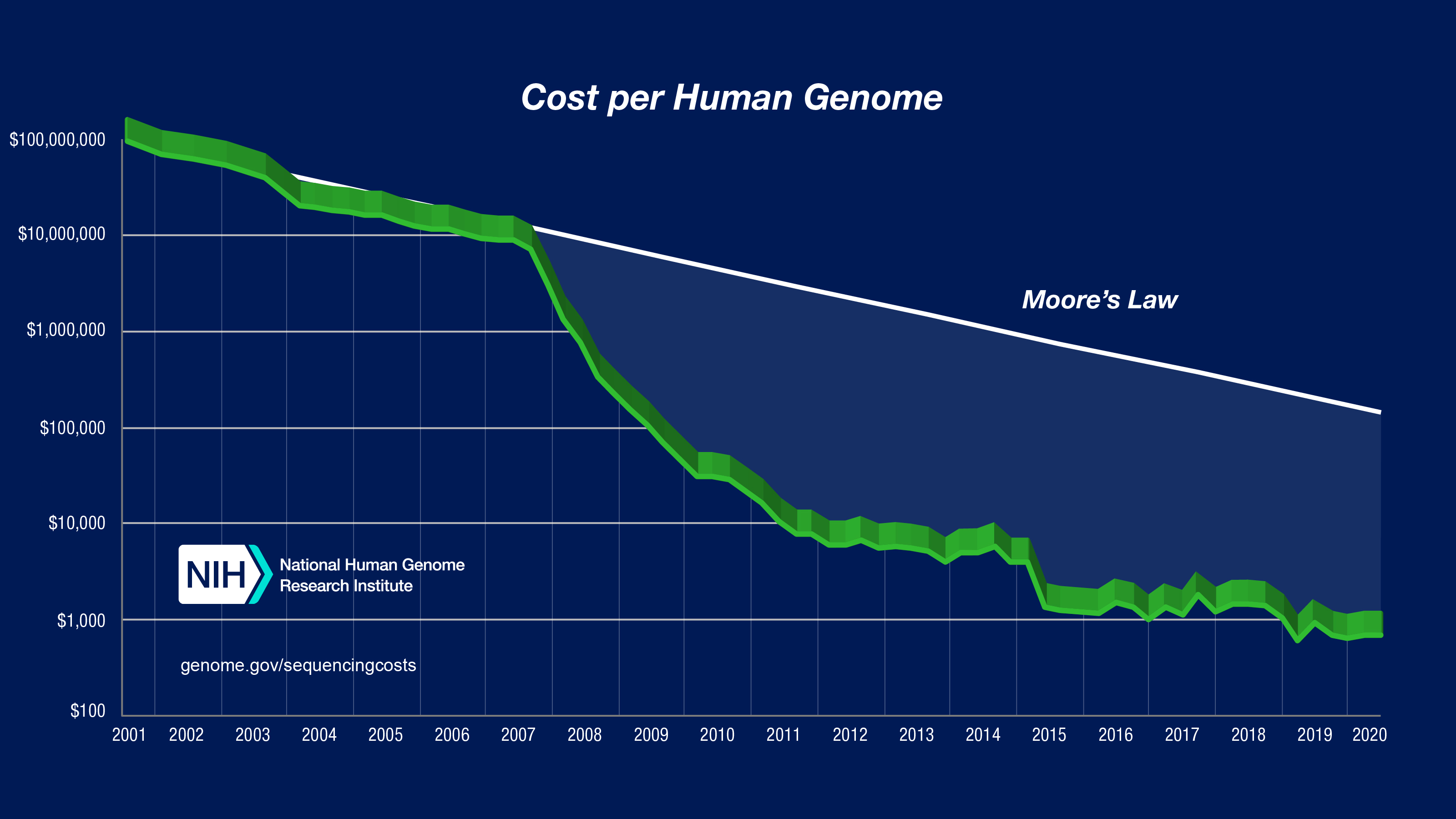
こちらは,アメリカ国立衛生研究所(National Institute of Health)が試算した一人分のヒトゲノムを読みとるのに必要な金額を示したグラフです.縦軸がコスト,横軸が年を示しています.現在の読み取り費用は約10万円ですが,20年前はなんと100億円必要だったのです.グラフを見ると,2000年代後半から急激にコストが下落しているのがわかると思います.一体,何が起きたのでしょうか?
実は,上述の「シークエンサー」に技術革新が起き,読み取り(シークエンシングと呼びます)コストが劇的に下がったのです.シークエンサーの性能向上は,生命科学の研究を大きく前進させました.そして同時に,同分野における計算機科学の重要性も大きく高めることになったのです.考えてみてください,個人の設計図が10万円で手に入るのです.世界中の研究機関では膨大なデータが算出されます.各設計図は30億塩基対に及ぶ大きさで,シークエンサーは設計図の断片を(多少のエラーを含めながら)大量,かつバラバラに読み出してくるのです.優れたアルゴリズムなくして解析を遂行することはできません.
シークエンサーの性能向上は日進月歩のため,その出力データを解析するアルゴリズムにも日進月歩の向上が求められています.D問題で使うデータは,Oxford Nanopore Technologies社のシークエンサーによるものです.こちらは,読み取り長がとても長いという優れた特徴を持ち,多くの解析の現場で用いられています.今回は,最新のキットQ20を用いて読み出したデータとなることにもご注目ください.
Score : 500 points
Problem Statement
s and t are strings consisting of
S_{sop} = \sum_{i=1}^{N} sim(s[i], t[i])
sim(x,y) is a function for calculating the similarity of x and y, and the following table defines its output.
| x \ y | |||||
| 1 | -3 | -3 | -3 | -5 | |
| -3 | 1 | -3 | -3 | -5 | |
| -3 | -3 | 1 | -3 | -5 | |
| -3 | -3 | -3 | 1 | -5 | |
| -5 | -5 | -5 | -5 | -5 |
For example when s=
S_{sop} = sim(
Note that s and t with balnk symbols are aligned such that same character appears at same position in each string for many positions.
C-TCGGT CATCC--
We call this task pairwise alignment and described it as strings in two lines.
Given two strings, output its pairwise alignment such that it maximizes S_{sop}.
Constraints
- s and t are strings consisting of
A ,C ,G ,T . - 10 \leq |s|, |t| \leq 400 (length of a string x is denoted by |x|.)
Input
Input is given from Standard Input in the following format:
s t
Output
Print a pairwise alignment of input strings such that S_{sop} is maximized.
After inserting
s should be printed in the first line, and t should be printed in the second line. If there is more than one alignment that maximizes S_{sop}, you can print any one of them.
The judging system can only accept user's output when:
- The first line/second line is equal to the first line/second line of the input if
Sample Input 1
AGTTGAATTT GTCGGACTTT
Sample Output 1
AGT-TGAATTT -GTCGGACTTT